INTRODUCTION
The First National Wilms' Tumor Study published in 1978 by Beckwith and Palmer [1] described a rare tumor pattern that was later referred to as a malignant rhabdoid tumor of the kidney (MRTK). Similar histological appearances were noted in both renal and extrarenal locations, including the central nervous system, lungs, mediastinum, peritoneal cavity, esophagus/stomach, duodenum, ileum, jejunum, colon, gallbladder, salivary gland, thyroid gland, uterus and other organs. The rhabdoid morphology has been described as occurring in epithelial tumors, as well as within various mesenchymal and biphasic malignancies, including synovial sarcomas [2], mixed epithelial and stromal tumors of the kidney [3] and melanomas [4]. We report a very rare occurrence of a rectal malignant rhabdoid tumor: few data are available on similar lesions in this particular location, with only two brief reports in the English literature having been published to date [5, 6].
CASE REPORT
A 49-year-old Caucasian male presented to the outpatient clinic with symptoms of tenesmus, abdominal pain, blood and mucus in stool, and weight loss of over 40 kg during the previous 6 months. A circular ulcerated and stenosing tumor was discovered on colonoscopy and was located 13 cm above the anal verge. A partial total mesorectal excision (TME) with a resection of the right ureter and urinary bladder (tumor penetrated these structures), as well as dissection of the right lateral pelvic node, was performed; 27 of 51 harvested lymph nodes contained metastases. No distant metastases or other tumors were discovered at the time of surgery.
Histopathology
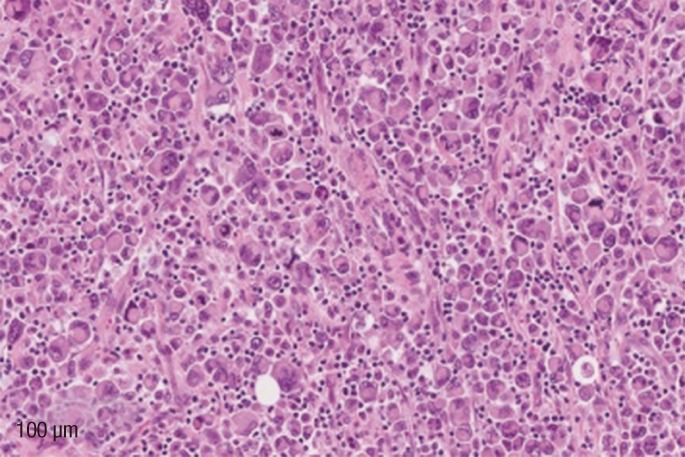
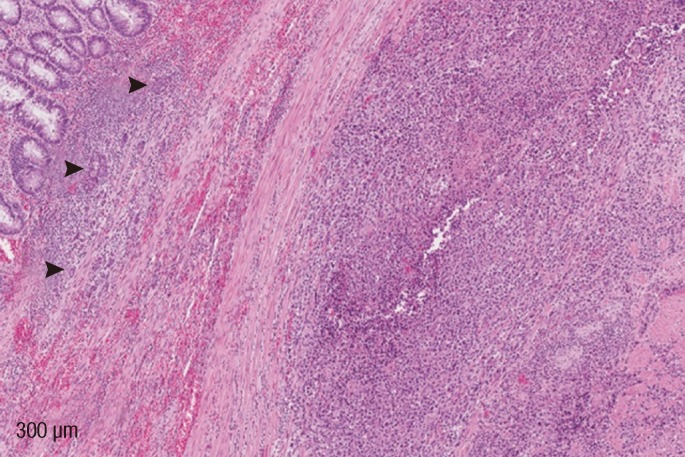
Gross examination demonstrated a broadly ulcerated, deeply infiltrative tumor within the rectal wall, measuring 7 cm in greatest dimension, growing through the adventitia into the adjacent urinary bladder that was removed together with a segment of bowel. Microscopically, the lesion was composed of two components: over 99% of it showed a rhabdoid morphology with discohesive solid sheets of pleomorphic epithelioid cells, little stroma and few inflammatory cells in between. Abundant eosinophilic cytoplasm with a large excentric nucleus and a single prominent nucleolus was present in the majority of the tumor cells. Numerous mitotic figures, including atypical forms, were present (Fig. 1). No "signet-ring" cells were identified. The second minor component constituted a poorly-differentiated adenocarcinoma and was present only in one tissue block section located at the periphery of the main tumor mass just underneath the intact colonic mucosa (Fig. 2). It was composed of irregular, cribriform glands lined by large atypical cells with prominent nucleoli and scant cytoplasm, surrounded by a desmoplastic inflammatory stroma.
Lymphovascular invasion was present. Metastatic disease was identified in multiple mesenteric lymph nodes and constituted a spread of pure rhabdoid cells without an adenocarcinoma component. Assessments of the surgical resection margins raised the possibility of microscopic residual disease.
Immunohistochemistry
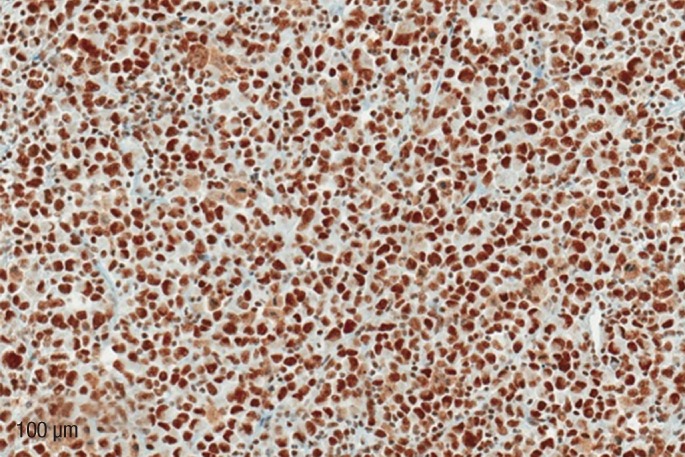
Rhabdoid cells demonstrated positivity for pancytokeratin, vimentin, and epithelial membrane antigen, and were nonreactive to cytokeratin 7 (CK7), cytokeratin 20 (CK20), thyroid transcription factor-1, homeobox protein CDX-2 (CDX-2), desmin and myogenic differentiation 1 (MyoD1). The minor adenocarcinoma component vanished on deeper sections; nevertheless, pancytokeratin, vimentin and CDX-2 stains were positive whereas CK7 and CK20 stains were both negative. INI1 antibody stained all neoplastic nuclei within the rhabdoid component (Fig. 3). A microsatellite analysis was undertaken by applying markers to MLH1, MSH2, MSH6, and PMS2 and showed no loss of expression of all 4 of them in tumor cells.
Molecular analysis
Mutation analysis of the rhabdoid tumor component revealed a wild-type (not mutated) KRAS gene and BRAF V600E mutation.
Differential diagnoses
Rectal lesions are strongly believed to represent a primary neoplasm based on their overall gross appearance and clinical presentation. They are circular, dominant rectal tumor with no other lesions being identified at the initial workup, making metastatic disease in the rectum almost impossible. The immunophenotypic pattern was informative with respect to conclusively confirming the rhabdoid morphology; however, it was not suggestive of organ specificity to help rule out secondary dissemination to the rectum. Identification of a minor adenocarcinoma component was most useful. It strongly argued for the lesion to represent a primary rectal composite rhabdoid tumor. With the rhabdoid morphology in mind, an epithelioid sarcoma was also on the differential list; however, given that both rhabdoid tumors and epithelioid sarcomas share almost identical histological, immunohistochemical and molecular patterns, separation of these two entities may have been impractical and nearly impossible [7]. Again, the presence of a minor adenocarcinoma component strongly favored a rhabdoid tumor, as it would not be expected to occur in an epithelioid sarcoma.
Clinical course
A month past surgery, a computed tomography (CT) scan disclosed multiple metastases within abdominal lymph nodes, iliac vessel invasion, and probable disease spread to the liver. Intensive chemotherapy was initiated: 4 cycles according to the chemotherapy regimen, consisting of folinic acid (leucovorin), fluorouracil (5-fluorouracil), and irinotecan (FOLFIRI) regimen were administered. The therapy course was complicated by development of bilateral pneumonia and ascites that were treated with antibiotics and drainage. Three months into the postoperative course, a repeated CT scan demonstrated disease progression despite aggressive chemotherapy. The patient received four additional cycles of chemotherapy according to the chemotherapy regimen, consisting of folinic acid (leucovorin), fluorouracil (5-fluorouracil), and oxaliplatin (FOLFOX4) regimen; however, again, no improvement was observed, metastatic disease rapidly spread to leptomeninges, neurological symptoms developed, and the patient eventually passed away 7 months after initial disease presentation.
DISCUSSION
The term "rhabdoid" is being applied to describe a histological pattern of a poorly-differentiated neoplasm composed of loosely cohesive polygonal cells with eccentric vesicular nuclei, prominent nucleoli, and eosinophilic cytoplasm that displaces the nucleus. These cells are characterized by aggregates of intermediate filaments found on electron microscopy and by a specific immunohistochemical staining pattern revealing combined positivity for vimentin and cytokeratins, together with an absent reactivity to muscle, vascular or melanocytic markers. Weeks et al. [8] outlined strict criteria for the definition of MRTK and proposed a hypothesis claiming that extrarenal rhabdoid tumors (ERRTs) represent a heterogeneous group of malignancies with a common phenotype, but no histogenic specificity. In ERRTs, the rhabdoid component may represent the sole histological pattern in so-called "pure" rhabdoid neoplasm or may comprise areas within epithelial, mesenchymal or other malignancies in what are defined as "composite" or "mixed" tumors [9, 10]. In composite lesions, the original component of the carcinoma or other malignancy defines the diagnostic category, with rhabdoid differentiation emphasized in the final report, as it signifies an aggressive biological behavior. Much controversy still exists as to whether ERRTs constitute a distinct nosological entity or whether they represent similarly appearing variants of de-differentiation within a primary malignancy at various localizations. There has been a focus on molecular genetics investigating changes in the tumor suppressor gene INI1 [11, 12] and the chromosome 22q [13] in rhabdoid lesions. INI1 (BAF 47) antibody serves as a surrogate marker for an intact INI1 gene and could easily be assessed immunophenotypically [12]. Investigators discovered very poor association of genetic alterations with ERRTs; intact INI1 expression was noted in the vast majority of lesions analyzed. Follow-up data in these cases, however, revealed an aggressive biological behavior, implying that the rhabdoid morphology by itself, even without INI1 gene abnormalities, signifies worse prognosis, poor response to conventional therapeutic modalities, and rapid disease progression [13]. INI1 gene mutations have been described in a number of different malignant neoplasms, including renal medullary carcinomas, epithelioid sarcomas, malignant peripheral nerve sheath tumors, etc. - a proof, that alterations of the INI1 gene are neither sensitive nor specific to rhabdoid tumors.
Published data on rhabdoid neoplasms arising in the lower gastrointestinal tract describe these malignancies as sharing many similarities in pathological appearance and clinical behavior to those from other locations. Of note, three investigators have searched for microsatellite instability, CpG island methylation, and KRAS and BRAF gene mutations in colonic rhabdoid tumors. Two of them reported that their published tumors harbored BRAF V600E mutation, wild-type KRAS and high methylation status [10, 14]; one case was reported to carry a wild-type KRAS gene and to have no evidence of microsatellite instability (BRAF gene and CpG island methylation status was not investigated) [15]. Similarly, our case demonstrated BRAF V600E mutation, wild-type KRAS and no evidence of microsatellite instability on immunohistochemical workup. However, we did not assess our case for CpG island methylation status. From the therapeutic standpoint, wild-type KRAS status would imply tumor responsiveness to novel biological molecules such as Bevacizumab or anti-epidermal growth factor receptor (EGFR) compounds; however, a BRAF mutation and rhabdoid morphology would signify the opposite: a poor response to conventional and novel chemotherapy regimens and a likely refractory disease with relapses. In line with the latter proposition was an observation reported by Pancione et al. [14], where despite multiple cycles of Bevacizumab and Cetuximab-based therapies, no significant benefit was noted, with rapid disease progression and short survival in their patient diagnosed with a rhabdoid tumor. Our patient shared a very similar fate: rapid tumor progression irrespective of intensive standard chemotherapy regimens (FOLFIRI and FOLFOX4); biological and anti-EGFR therapies were not attempted due to clinical contraindications.
This case illustrates the importance of correct classification and identification of particular rare components within neoplastic lesions: histological identification of rhabdoid morphology was an early and accurate predictor of poor outcome despite available therapies. Future studies, especially on the biology and the genetics of these tumors, are important to search for specific therapeutic agents and more effective treatment regimens.