Effects of DCA on Cell Cycle Proteins in Colonocytes
Article information
Abstract
Purpose
Evidence that indicates bile acid is a promoter of colon cancer exists. Deoxycholic acid (DCA) modifies apoptosis or proliferation by affecting intracellular signaling and gene expression. However, because previous studies have been based on studies on colon cancer cell lines, the effect of DCA on normal colonocytes is unknown.
Methods
Normal colonocytes and Caco-2 and HCT116 cells were treated with 20 µM and 250 µM of DCA, and the effect of different concentrations of DCA was measured based on the expression of cell-cycle-related proteins by using Western blots.
Results
The expressions of CDK2 and cyclin D1 for different concentrations of DCA in normal colonocytes and colon cancer cells were similar, but the expressions of cyclin E and A were significantly different. In HCT116 colon cancer cells, the expression of cyclin E increased regardless of the DCA concentration, but in normal colonocytes and Caco-2 cells, the expression of cyclin E was not changed or decreased. In HCT116 colon cancer cells, the expression of cyclin A was not changed or decreased regardless of the DCA concentration, but in normal colonocytes and Caco-2 cells, the expression of cyclin A was increased at a DCA concentration of 20 µM.
Conclusion
The effect of DCA on stimulating cell proliferation suggests that DNA synthesis is stimulated by an increased expression of cyclin E in colon cancer cells. Our results suggest that a low dose of DCA induces cellular proliferation through increased expression of cyclin A and that a high dose of DCA induces decreased expression of cyclin E and CDK2 in normal colonocytes.
INTRODUCTION
Epidemiological investigation found that an increase in fecal cholic acid is a risk factor and facilitates the development of colon cancer [1, 2]. Also, increased levels of cholic acid concentration in the stool and blood were found in patients with a large intestine adenoma. These results indicate that an increased level of cholic acid in the stool has a relationship with colon cancer development [1]. However, the strongest proof that cholic acid is related with colon cancer development can be found in the animal model for treating a tumor in the large intestine with azoxymethane (AOM). The group that had food with added deoxycholic acid (DCA) showed a more tumor than the control group with just AOM treatment [2-4]. In addition, by changing the flow of cholic acid of a mouse through operation and treating with AOM, Morvey [5] reported that large intestine, which was not exposed to cholic acid, showed very few or almost no tumors when compared with the large intestine exposed to cholic acid.
Secondary bile acid, such as DCA, is well known to be a tumor facilitating factor that will induce exfoliation of epithelial cells from the surface of the large intestine mucosa when present in concentrations of 2-5 mM. Through a compensatory mechanism, hyperplasia of large intestine epithelial cells will occur and facilitate tumor development [6, 7]. According to other studies, a low concentration of secondary bile acid increases the number of aberrant crypt foci (ACF) [3] in the large intestine, and DCA in concentrations greater than 50 µM will cause colon cancer cell death. The studies demonstrated that, according to these effects of DCA in low concentrations, cell death caused by cholic acid will play a crucial role in facilitating tumor development in the large intestine [8]. However, the mechanism explaining how DCA or other cholic acids act is not yet clearly understood. There are reports about the tumor-facilitating effect affecting intracellular signal transduction and gene expression and eventually causing cell death or altered cell proliferation.
DCA activates AP-1, and activation of protein kinase C (PKC) and mitogen-activated protein kinase (MAPK) is necessary for AP-1 activation [9]. MAPK is an important signal transduction factor that delivers various extracellular stimuli to the inside of the cell, which controls cell proliferation, differentiation, and death. Most of the transportation of extracellular signals to the nucleus at the cell membrane is done by three types of MAPKs: extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAP kinase (p38). Although DCA activates both ERK and p38, only the ERK activated by DCA affects cell death caused by DCA. However, there are reports that have showed that increased activation of ERK by DCA suppressed cell death caused by DCA [10]. These reports convey the ideas that DCA has the function of stimulating cell hyperplasia and cell death and that high concentrations of DCA generally induce cell death. However, the mechanism for or the cause of DCA inducing cell hyperplasia or cell death is still unknown.
In order for cholic acid to induce large-intestine epithelial-cell deformation or cancer development, the epithelial cell must be exposed to cholic acid for a long period of time. Prevention of cancer caused by cholic acid would be possible if the mechanism of a normal mucosa epithelial cell turning into cancer is discovered. Since the researchs until now used colon cancer strains, it was difficult to say that DCA reflected the action of a normal large-intestine mucosa epithelial cell. Thus, this research was conducted by separating and culturing normal large-intestine mucosa cells. The normal large-intestine mucosa cells were then compared with cells of the Caco-2 colon cancer strain (containing the characteristics of normal cells) through a study of the variations in cell-cycle protein expression with changing DCA concentration. The results were compared with the expressions from the HCT116 colon cancer cell strain to search for the effect of DCA on the protein-expression-related cell cycle.
METHODS
Materials and antibodies
The antibodies used in this experiment were CDK2 (M2, 1 : 1,000, anti-goat polyclonal antibody against a peptide mapping at the carboxy terminus of CDK2), cyclin D1 (C-20, mAb), cyclin E (HE-12, mAb and C-19 pAb), and cyclin A (C-19), which were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The culture medium used in the cell culture was RPMI1640 (JEIL Biotech Services Incorp, Daegu, Korea), and fetal bovine serum (FBS, Hyclone Laboratories, Logaar, UT, USA), penicillin-streptomycin, and deoxycholate (Sigma, St Louis, MO, USA) were added reagents. The PVDF membrane, the SDS-PAGE electrophoresis apparatus, and the semi-dry membrane transfer equipment were purchased from Bio Rad (Hercules, CA). All other reagents were purchased from Sigma Biochemical. The HCT116 colon cancer cell strain and the Caco-2 colon cancer cell strain, which is known to have normal large-intestine mucosa-cell properties, were purchased from the Korean Cell Line Bank.
Separating and culturing normal large intestine mucosa cells
Samples of normal large-intestine mucosas were collected during surgery for colon cancer or benign large-intestine disease, and the samples were collected at least 5 cm from the lesion site. The samples were separated with caution in order to prevent muscle cells underneath the mucosa and the submucosal layer from being included. Blood vessels in the submucosal layer and connective tissue were removed from the sample. The mucosal sample was then immediately placed in 50 mL of chilled, ice cold, Hank's Balanced Salt Solution (HBSS, 100 U/mL PCN-SM, 50 µg/mL GM, 2.5 µg/mL Amphotericin B, without Ca+2, Mg+2). Lipid and necrotic tissue were removed and the sample was cut into 3- to 4-mm-sized strips. Then, the samples were washed with HBSS several times. Fifty mL of HBSS (1 mM DTT, 1 mM EDTA, 100 U/mL PCN-SM, 50 µg/mL Gentamicin, 2.5 µg/mL Amphotericin B, without Ca+2, Mg+2) was placed in a beaker, and magnetic stirring bar was used at room temperature for 30 minutes at a speed of 0.3 g to remove the remaining mucous and sludge. The stirring speed was controlled in order for all the epithelial cells to float in the fluid. After cleaning the epithelial cells with HBSS, the sample was once more stirred and reacted with 100 mL of HBSS (1 mM EDTA, 100 U/mL PCN-SM, 50 µg/mL Gentamicin, 2.5 µg/mL Amphotericin B, without Ca+2, Mg+2) without DTT at room temperature for 60 minutes. Supernatant-containing crypts were collected at least twice, and the crypts were set on top of ice to let them settle quickly. Collected crypts were washed with RPMI culture fluid containing the same antibiotics. Then, a suspension containing 15 mL of RPMI culture fluid and 10% fetal bovine serum was created before culturing. Two hundred U/mL of collagenase (Sigma type I) and 270 U/mL of DNase were added to the separated crypt samples and were reacted for 60 minutes at 37℃. Separated large-ntestine epithelial cells were cultured in RPMI culture fluid containing 10% fetal bovine serum.
Deoxycholic acid treatment
The concentration of DCA in the experiment was determined by using the MTS method, and the survival rates of the HCT116 colon cancer strain were measured when different concentrations of DCA were added and cultured for 3 days. Twenty µM and 250 µM were used as concentration values. Separated normal large-intestine epithelial-cell samples were treated with DCA in different concentrations and were cultured for 24 hours. As a control group, one sample was cultured for 24 hours without adding DCA. The Caco-2 strain and the HCT116 colon cancer strain were cultured in the same way to compare their results with those for normal large-intestine epithelial cells.
Measuring expression of proteins related to the cell cycle by using western blots
The cell samples were cultured for 24 hours (72 hours for the Caco-2 strain) at each concentration of DCA and were washed with cooled 0.1 M PBS (8.5 mM Na2HPO4·7H2O, 3 mM KH2PO4, 125 mM NaCl) 3 times. Cells were dissolved in cell lysis buffer solution (25 mM Tris-Cl [pH 7.0], 1 mM EDTA, 1 mM PMSF, 1 mg/mL leupeptin, antipain, aprotinin 0.1% NP-40, 250 mM NaCl, 5 mM DTT, 0.1% Triton-X-100). Cell homogenate went through the centrifuge at 12,000 rpm for 15 minutes at 4℃. Then, the supernatant was separated and stored at -80℃ before being used. Protein quantification was measured using the Bradford micro-assay method.
Thirty µg of cell lysate was added to each Laemmli sample buffer (0.5 M Tris-HCl [pH 6.8], 10% glycerol, 2% [w/v] SDS, 5% [v/v] 2-β-mercaptoethanol and 0.05% bromophenollue) and was degenerated at 95-100℃ for five minutes by boiling. Each of the specimens went through electrophoresis in 10% polyacrylamide gel by using the Laemmli method. Separated proteins were moved to a nitrocellulose membrane by using the semi-dry transblot apparatus. This membrane was placed in a solution with 5% non-fat skim milk dissolved in 1% TBS with 0.1% Tween-20 buffer (Tris 50 mM, 250 mM NaCl, 0.1% Tween-20, pH 7.5) was allowed to sit overnight at 4℃. The primary antibody mentioned above was dissolved in 1% TBS with 0.1% Tween-20 buffer solution overnight in the ratio of 1 : 1,000 at 4℃. After the primary antibody reaction, the sample was washed three times in 1% TBS-T for 10 minutes each. Then, it was reacted with peroxidase-conjugated secondary antibody (anti-mouse for monoclonal antibody, anti-goat for polyclonal antibody, 1 : 2,500 dissolved, Zymed Laboratories Inc, San Francisco, CA, USA) for 1 hour at room temperature. It was then re-washed three times with 1% TBS-T for 10 minutes each wash. Remaining immunoreactive protein was reacted by using a chemiluminescence kit (ECL kit, Amersham Co. Uppsala, Sweden) and following the manufacturer's directions, and immunological reaction strips were detected with an image analyzer (LAS-2000, Fuji, Tokyo, Japan). ERK protein expression was used as a control for internal protein expression.
RESULTS
When a 50 µM, 200 µM or 250 µM concentration of DCA was added to the HCT116 colon cancer cell strain and cultured for 72 hours, at the 200- and 250 µM concentrations, colon cancer strain proliferation was suppressed. However, for 50 µM of DCA, the proliferation of the strain was increased when compared with the control group without DCA added. In deciding the concentration of DCA, we used the MTS method to measure the toxicity of DCA. There was no cytotoxicity until the concentration of DCA reached 100 µM. In concentrations greater than 250 µM, approximately half of the colon cancer cells showed cytotoxicity when cultured for 72 hours. Although normal large-intestine epithelial cells should be used to measure the effects of DCA (cell proliferation and cytotoxicity), the effects were impossible to measure after culturing for 72 hours because 50% of the normal large-intestine epithelial cells survived after culturing for 24 hours without adding DCA. As an alternative, HCT116 colon cancer cells were used to determine the effects of DCA in different concentrations, and the results were used as standards. Twenty µM DCA was used as a low concentration, and 250 µM was used as a high concentration. Each was cultured for 24 hours, and changes in cell-cycle-related protein expression caused by DCA were measured.
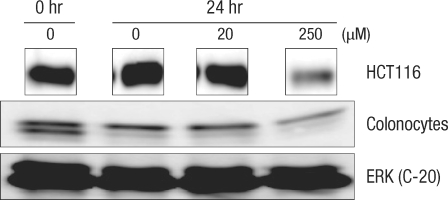
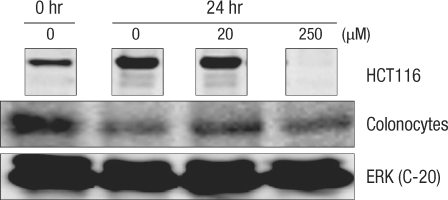
When normal large-intestine epithelial cells (Fig. 1) were cultured for 24 hours, a prominent decrease in the number of cells was found even without adding DCA. A similar cell count decrease was measured in the colon cancer cell group with 20 µM DCA. Even in colon cancer cells where approximately 50% of the cells showed cytotoxicity, most of the large-intestine epithelial cells showed cell death when 250 µM of DCA was added (Fig. 1).

Isolated human colonic crypt (A) and colonocytes (B). Human normal colonocytes were incubated for 24 hours with various concentration of deoxycholic acid: 0 (C), 20 µM (D), and 250 µM (E).
After culturing for 24 hours, expression of cell-cycle-related protein was measured by using the Western blot method (Figs. 2 and 3). In normal large-intestine epithelial cells, independent of the DCA concentration, there was no change in cyclin D1 expression. No cyclin D1 expression was found in Caco-2 cells. Expression of CDK2 did not change in the 20 µM DCA group, but there was a decrease in the 250 µM DCA group. The same expression pattern was found in Caco-2 cells. Expression of cyclin E did not show any change after culturing for 24 hours, independent of the concentration of DCA. In Caco-2 cells, no noticeable change of expression was measured when 20 µM of DCA was added, but a significant decrease was measured when 250 µM of DCA was added. Expression of cyclin A was increased for a concentration of 20 µM, but did not change when 250 µM of DCA was added. Caco-2 cells, however, showed the same pattern as that shown by normal large-intestine epithelial cells (increased expression at 20 µM), but no expression was measured at 250 µM of DCA.

Change of cell-cycle-related proteins in human colonocytes after incubation for 24 hours with deoxycholic acid based on a Western blot analysis. Conc., concentration; ERK, extracellular signal-regulated kinase.
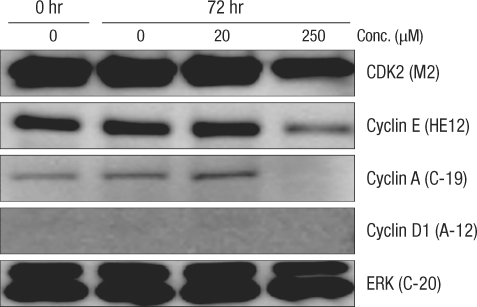
Comparison of the change in cell-cycle-related proteins in the Caco-2 colorectal cancer cell-line after incubation for 72 hours with deoxycholic acid based on a Western blot analysis. Conc., concentration; ERK, extracellular signal-regulated kinase.
Expressions of cell-cycle-related protein in normal large-intestine epithelial cells with DCA are compared with expressions in HTC116 colon cancer cells (Figs. 4-7). In colon cancer cells, an increase of cyclin E expression was measured in groups with added DCA independently of the DCA concentration when compared with the group with no DCA added. However, no change in expression was found in normal large-intestine epithelial cells, and this result was independent of the DCA concentration (Fig. 4). CDK2 expression in colon cancer cells was slightly reduced or did not change; showing the same results as those for normal large-intestine epithelial cells or Caco-2 cells (Fig. 5). There was no change in expression of the cyclin A protein in colon cancer cells cultured for 24 hours in 20 µM DCA. However, no expression was found when 250 µM DCA was added. In the case of normal large-intestine epithelial cells, an increase of cyclin A expression was measured for 20 µM DCA, but no change in expression was measured for 250 µM DCA (Fig. 6). Expression of cyclin D1 protein in colon cancer cells did not change when 20 µM DCA was added, but no expression was measured when 250 µM DCA was added. However, normal large-intestine epithelial cells did not show any change in the expression of cyclin D1, independent of the DCA concentration (Fig. 7).

Comparison of the change in the cyclin E protein in human colonocytes and in HCT116 colon cancer cells after incubation for 24 hours with deoxycholic acid based on a Western blot analysis. ERK, extracellular signal-regulated kinase.

Comparison of the change in the cyclin D1 protein in human colonocytes and HCT116 colon cancer cells after incubation for 24 hours with deoxycholic acid based on a Western blot analysis. ERK, extracellular signal-regulated kinase.
Comparison of the change of Cdk2 protein after incubation for 24 hours with deoxycholic acid in human colonocytes and HCT116 colon cancer cells by Western blot analysis. ERK, extracellular signal-regulated kinase.
Comparison of the change in the cyclin A protein in human colonocytes and HCT116 colon cancer cells after incubation for 24 hours with deoxycholic acid based on a Western blot analysis. ERK, extracellular signal-regulated kinase.
DISCUSSION
Cholic acid, a derivative of cholesterol, is necessary for controlling transcription of the gene that controls dietary fat absorption and cholesterol homeostasis. However, cholic acid can induce various biological effects depending on its different chemical structures [8]. Ursodeoxycholic acid (UDCA), one of the hydrophilic cholic acids, protects and prevents cell death caused by other hydrophobic cholic acids [11]. DCA was chosen from among the various kinds of cholic acids for this experiment because DCA is known to have the strongest cytotoxicity [12].
For low concentrations of DCA (< 50 µM), cell proliferation of the HCT116 colon cancer strain was stimulated whereas concentrations greater than 200 µM induced cell death as the cells were detached from the culture flask, thus showing the two different effects (cell proliferation and cell death) that DCA has, depending on the concentration. According to other research, these effects of DCA on colon cancer strains were also found in the Caco-2 strain (maintaining fetus' normal large-intestine mucous cells) [13] and the HT-29 colon cancer strain (undifferentiated cancer cells) [14]. These results match the data showing that the colon cancer strain proliferates in DCA lower than 5 µM [15] and dies in DCA higher than 50 µM [8]. According to the cytotoxicity measurements done with the MTS method, the cell survival rate decreases greatly when the DCA concentration is greater than 100 µM, thus showing significant cytotoxicity. This measurement of the DCA concentration, however, was done using the colon cancer strain. For an accurate experiment, normal large-intestine epithelial cells must be used for testing cell proliferation and cytotoxicity caused by DCA. However, because of the low survival rate of normal large-intestine epithelial cells (< 50% when cultured for 24 hours without DCA), testing cytotoxicity was impossible to measure after culturing for 72 hours. Thus, HCT116 colon cancer cells were used to set the standard for DCA concentration. Twenty µM and 250 µM were considered as the low concentration and the high concentration of DCA, respectively. Cells were cultured for 24 hours, and changes in cell-cycle-related protein expression due to DCA were measured.
DCA showed four large-intestine epithelial-cell proliferation effects: 1) increased mutation of the k-ras gene in large-intestine tumors [16], 2) increased expression of AP-1 (human colon cancer cell transcription factor) [9], 3) suppressed cell death caused by butyrate in cultured neoplastic large-intestine epithelial cells [17], and 4) directly stimulated colon cancer cell movement [18]. However, a high concentration of DCA induces cell death in large-intestine cells [8-10]. All these effects are probably caused by DCA controlling the intercellular signal transduction system through PKC activation of the phobol ester effect. Various kinds of cholic acids also suppress cell proliferation and cell death in breast cancer cells. They also reduce expression of Bcl-2 and cyclin D1, but are reported to not have an effect on CDK2 and CDK4. Also, these effects on the cell cycle are reported to be independent of p53 expression [19].
According an experimental result, CDK2 protein showed the same expression changes in normal large-intestine epithelial cells, the Caco-2 strain, and the HCT116 colon cancer strain when different concentrations of DCA were added. Cyclin D1 either did not show any expression change or showed a slight decrease. Expression changes caused by DCA concentration between the HCT116 colon cancer strain and the normal large-intestine epithelial cells were especially noticeable for cyclin E and cyclin A. Cyclin E expression was increased in the HCT116 colon cancer strain, regardless of the concentration of DCA. On the other hand, it was either decreased or did not change in normal large-intestine epithelial cells and Caco-2 cells. Cyclin A protein expression showed either no change or a decrease in HCT116 colon cancer cells whereas an increase in expression was found in normal large-intestine epithelial cells and Caco-2 cells at 20 µM.
The proliferation effect of DCA did not include increased expression of cyclin D1 (curator of cell-cycle entrance from the G1 phase). In colon cancer cells, cyclin E (necessary for G1→S phase change) expression was increased, stimulating cell DNA fusion. However, in normal large-intestine epithelial cells, DCA did not show any effect on cyclin E expression. DCA also did not affect the expression of CDK2, which combines with cyclin E and becomes a phosphorylase. In normal large-intestine epithelial cells, a low concentration of DCA increases expression of cyclin A (combines with CDK2 and CDK1 of the S phase and becomes a phosphorylase) and causes cell proliferation. At a high concentration of DCA, CDK2 expression and activation of CDK2 phosp horylase are reduced, suppressing cell proliferation.
In large-intestine cancer cells, DCA increases the expression of cyclin E (necessary for entering the S phase from the G1 phase), which facilitates DNA synthesis and stimulates cell proliferation. On the other hand, a low concentration of DCA in normal large-intestine epithelial cells shows increased expression of cyclin A, which activates phosphorylase by fusing with S phase CDK2 and S phase CDK1, thus showing a cell proliferation effect. For high concentrations of DCA, cycline E, which is essential to going from the G1 phase to the S phase and to the expression of activated CDK2 combined with cycline E, is suppressed, and activation of CDK2 phosphorylase is reduced, thus suppressing cell proliferation.
Notes
No potential conflict of interest relevant to this article was reported.