

INTRODUCTION
Prostaglandin (PG) is a bioactive lipid compound produced from arachidonic acid. Specific forms of PGs, including PGE2, PGD2, PGF2a, PGI2, and thromboxane A2 play crucial roles in physiology, including renal function maintenance, gastrointestinal function maintenance, regulation of vascular homeostasis, and in pathological conditions like the inflammatory response [1, 2]. Among different PGs, PGE2 is specifically known to be related to various stages of cancer progression. Cyclooxygenase (COX) plays the most significant role in PGE2 production. There are primarily 2 types of COX. COX-1 is involved in normal physiological function by maintaining a constant amount. And inducible COX-2 expression is increased by stimulation such as cytokines and other inflammatory mediators [2]. COX-2 is also known for its role as an oncogene and is overexpressed in various solid tumors including colorectal cancer [3, 4]. Pharmacological inhibitors of COX, such as aspirin, sulindac, and celecoxib have been shown to lower the incidence of colon cancer, gastric cancer, and esophageal cancer. Moreover, the mortality rate of these cancers has declined based on results of epidemiologic studies [5]. Among enzymes that play a crucial role in PGE2 synthesis along with COX-2, there is prostaglandin E synthase (PGES), which switches COX-induced PGH2 to PGE2. Cytosolic PGES is known to be involved in the production of PGE2 and is primarily associated with COX-1. Microsomal PGES-1 (mPGES-1) is involved in PGE2 synthesis mediated by COX-2. Recent studies indicate that mPGES-1 is overexpressed in colon cancer and other various types of cancer, including lung and stomach cancer, and is known to have an important role in tumorigenesis [6, 7]. Additionally, 15-prostaglandin dehydrogenase (15-PGDH) functions as an enzyme to break down PGE2, and as a tumor suppressor. Furthermore, expression of 15-PGDH is suppressed in various tumors [8] and previous studies conducted by the authors confirmed the inhibition of 15-PGDH expression in colon cancer [6]. Taken together, the effects of these enzymes on cancer can ultimately be explained by the effects of PGE2. It is reported that generation of PGE2 plays an important role in promotion of tumor growth and development, resistance to apoptosis, proliferation, invasion and metastasis, angiogenesis and drug resistance in colon cancer [9]. Despite the important role of PGE2, there are few studies directly measuring PGE2 in colon cancer.
The current study was designed to investigate the expression of PGE2 in patients with colorectal cancer and in colon cancer cells and to investigate the changes in the expression of PGE2 related enzymes upon induction of PGE2.
METHODS
Patients
This study was carried out following approval by the Institutional Review Board of Chosun University Hospital (No. IRB-11S-225). A total of 30 patients pathologically diagnosed with colorectal adenocarcinoma who had undergone curative resection were enrolled in the study upon completion of informed consent. Following removal of tumor-bearing specimens, 3 to 4 small pieces of samples were evenly excised from the cancer region, along with 3 to 4 small pieces that were collected from the seemingly normal mucosa of the colon located at least 3 to 4 cm away from the tumor. These tissues were thoroughly cleaned in normal saline and stored in liquid nitrogen at ŌĆō80┬░C for further analysis. Among the patients enrolled, 26 had PGE2 both in the tumor tissues and the adjacent mucosa sufficient enough to be measured. This group was comprised of 19 patients with colon cancer and 7 with rectal cancer. The mean age was 62.8 years, 5 of them were in TNM stage I, 13 were in stage II, and 8 were in stage III. Of the 26 patients, 2 were taking aspirin regularly and 3 were taking other nonsteroidal anti-inflammatory drugs intermittently. Upon completion of tissue sampling, 100 mg of cancer tissue and 100 mg of normal mucosal tissue of each patient were homogenized using a sonicator and separated into 2 parts. After being left for 30 minutes at 4┬░C the supernatant was rapidly stored at ŌĆō80┬░C and later used for calculation of total protein. The second part of the homogenate was used for the isolation of PGE2.
Materials
The antibodies against total actin, COX-1, and COX-2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse monoclonal antibody for mPGES-1 and PGE2 enzyme immunoassay kit was obtained from Cayman Chemical (Ann Arbor, MI, USA). Rabbit polyclonal antibody to 15-PGDH was obtained from Novus Biologicals (Littleton, CO, USA). Aspirin, meloxicam, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), tumor necrosis factor (TNF)-╬▒, and pyrrolidine dithiocarbamate (PDTC) were obtained from Sigma/Aldrich (St. Louis, MO, USA). Mouse monoclonal antibody to nuclear factor kappa B (NF-╬║B), inhibitor of NF-╬║B (I╬║B) were obtained from Santa Cruz Biotechnology.
PGE2 assay
PGE2 was measured with an enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturerŌĆÖs instructions. For measurements of the concentration of PGE2, cancer cells were immersed in culture medium, whereas cancer tissue/normal mucosa tissue samples were homogenized. Briefly, 1 mL of ethanol was added to 250 ┬ĄL of the homogenated tissue samples, vortexed, incubated at 4┬░C for 5 minutes, and centrifuged at 3,000├Śg for 10 minutes at 4┬░C to remove proteins. The supernatant was collected, ethanol was evaporated by centrifugation, and acetate buffer (pH= 4.0) was added to acidify the sample to a pH -4.0. Next, 5 mL of methanol (1%) and ethyl acetate were added to each sample, and PGE2 was extracted by passing through solid phase extraction cartridge (C18) columns. After evaporation of ethyl acetate and methanol, ELISA buffer was added and PGE2 concentration was measured by ELISA. All samples were measured in triplicate. Positive control was used to create a standard curve consisting of 8 serial dilutions, ranging from 1,000 to 10 pg/mL of PGE2.
Cell culture
HCA-7 was cultured in DulbeccoŌĆÖs Modified Eagle Medium supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic antimycotic (penicillin of 100 U/mL and streptomycin of 50 ┬Ąg/mL). HT-29 cells were cultured in MacoyŌĆÖs 5A supplemented with 10% FBS and 1% antibiotic antimycotic. FET cells were cultured in Modified Eagle Medium supplemented with 10% FBS and 1% antibiotic antimycotic.
Western blot analysis
Cells were disrupted, and the supernatant fractions were boiled for 5 minutes. The protein concentration was determined using a dye-binding protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA) as described in the manufacturerŌĆÖs manual. Lysate protein was subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to a polyvinylidene fluoride membrane. After blocking, the membrane was incubated with the appropriate specific primary antibody at 4┬░C overnight. Protein bands were visualized using a chemiluminescence detection kit after hybridization with the appropriate horseradish-peroxidase-conjugated secondary antibody.
Cell viability assay
Cell viability was measured using the MTT assay. Briefly, FET cells (2├Ś 104) were seeded into 96-well plates and cultured for 24 hours. The cells were treated with aspirin and meloxicam combined with TNF-╬▒ at various concentrations. The cells were cultured at different time points at 37┬░C, followed by incubation with MTT for 4 hours. The optical density of each well was measured at 570 nm using an ELISA reader (EMax, Molecular Devices, San Jose, CA, USA).
RESULTS
Expression of PGE2 and its related enzymes in patients with colorectal cancer
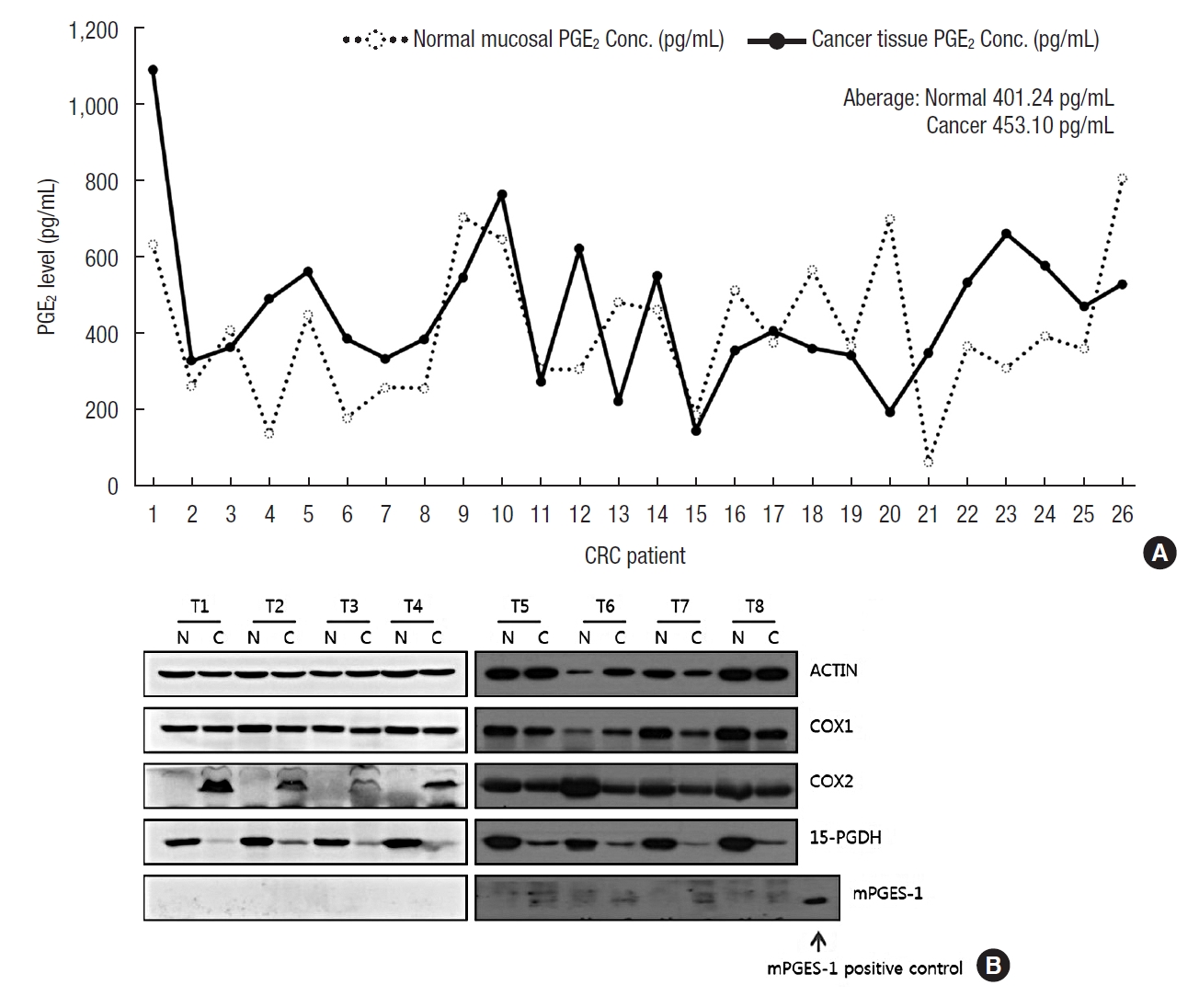
The mean level of PGE2 in the normal mucosa and cancer tissues was 401.2 and 453.1 pg/mL respectively, with no significant difference (P=0.247) (Fig. 1A). In 16 patients, the level of PGE2 was increased in cancer tissues compared to the normal mucosa, but in the remaining 10 patients, PGE2 in cancer tissues was decreased. No characteristic findings were observed in PGE2 levels in the patients taking nonsteroidal anti-inflammatory drugs. In terms of the expression of PGE2-related enzymes, COX-1 was expressed relatively evenly in the cancer tissues as well as normal mucosal tissues. COX-2 was consistently expressed in the cancer tissues, but not in the normal mucosal tissues. mPGES-1 was weakly expressed in some tissues but was not observed in the majority of the cancer and normal mucosal tissues. 15-PGDH was expressed in the normal mucosal tissues, while its expression was attenuated in the cancer tissues (Fig. 1B).
Basal expression of COX-2, mPGES-1, and other related enzymes in colon cancer cells
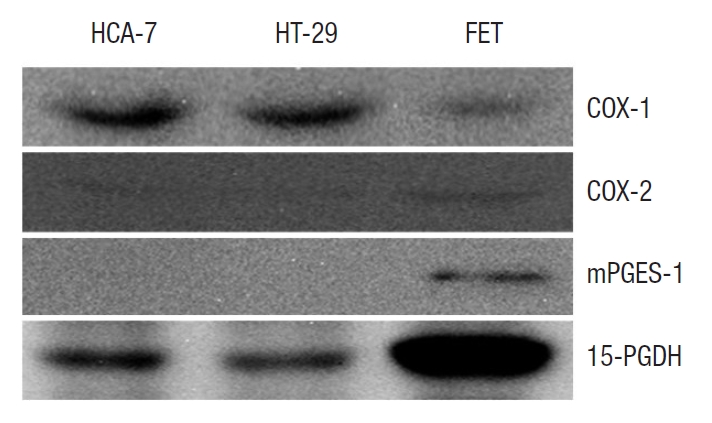
Basal expression of PGE2-related enzymes was examined in 3 different colon cancer cells; HCA-7, HT-29, and FET, using the Western blot assay (Fig. 2). COX-1 was present in all cells, whereas COX-2 and mPGES-1 were expressed only in FET cells. 15-PGDH was observed in all cells, but most frequently in FET. Given the increased PGE2-related enzymes in FET cells, additional experiments were conducted to explore these results.
Changes in PGE2 levels according to the TNF-╬▒ and aspirin in FET colon cancer cells
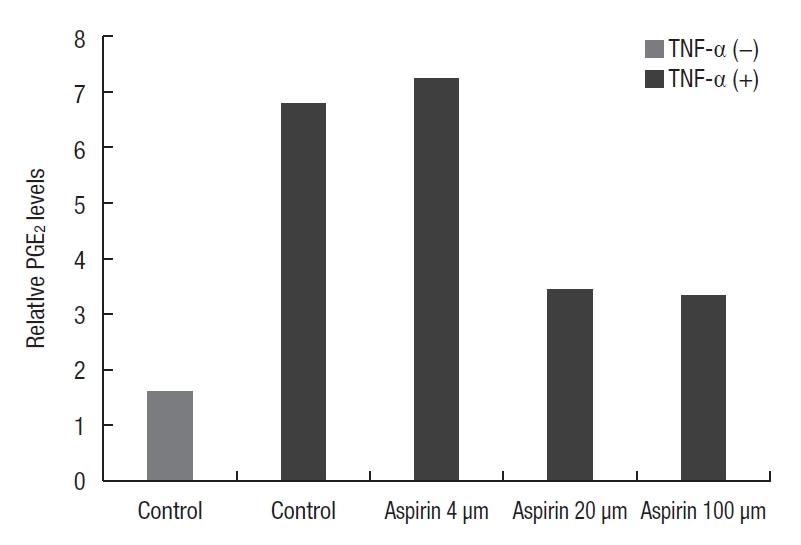
Changes in the PGE2 levels were observed in the following 3 cases: when PGE2 was in its basal state in FET cells; after TNF-╬▒ of 10 ng was administered for 12 hours; and after aspirin, a COX inhibitor, was administered. It was confirmed that PGE2 was upregulated in FET cells by stimulation of TNF-╬▒, and PGE2 elevation was inhibited by aspirin (Fig. 3). This demonstrated that PGE2 was actively metabolized in FET cells, which is related to the activity of PGE2-related enzymes.
Effect of TNF-╬▒ on the expression of PGE2 related enzymes in FET cells
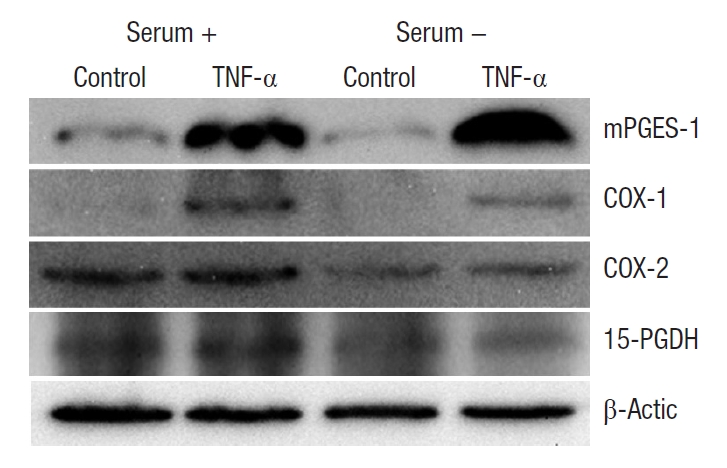
Several enzymes relevant to the metabolism of PGE2 were measured by Western blot after administering 10 ng/mL of TNF-╬▒ for 12 hours (Fig. 4). The experiment was conducted under the same conditions as stated previously, employing the serum-free media to block the effects of various substances contained in the cell culture media. Upon the administration of TNF-╬▒, mPGES-1 was found to significantly increase. COX-1 was also slightly elevated. However, COX-2 and 15-PGDH showed no difference even after the injection of TNF-╬▒. The experiment using the serum-free media demonstrated the same findings. These results suggest that elevated expression of PGE2 triggered by TNF-╬▒ is primarily due to the induction of mPGES-1.
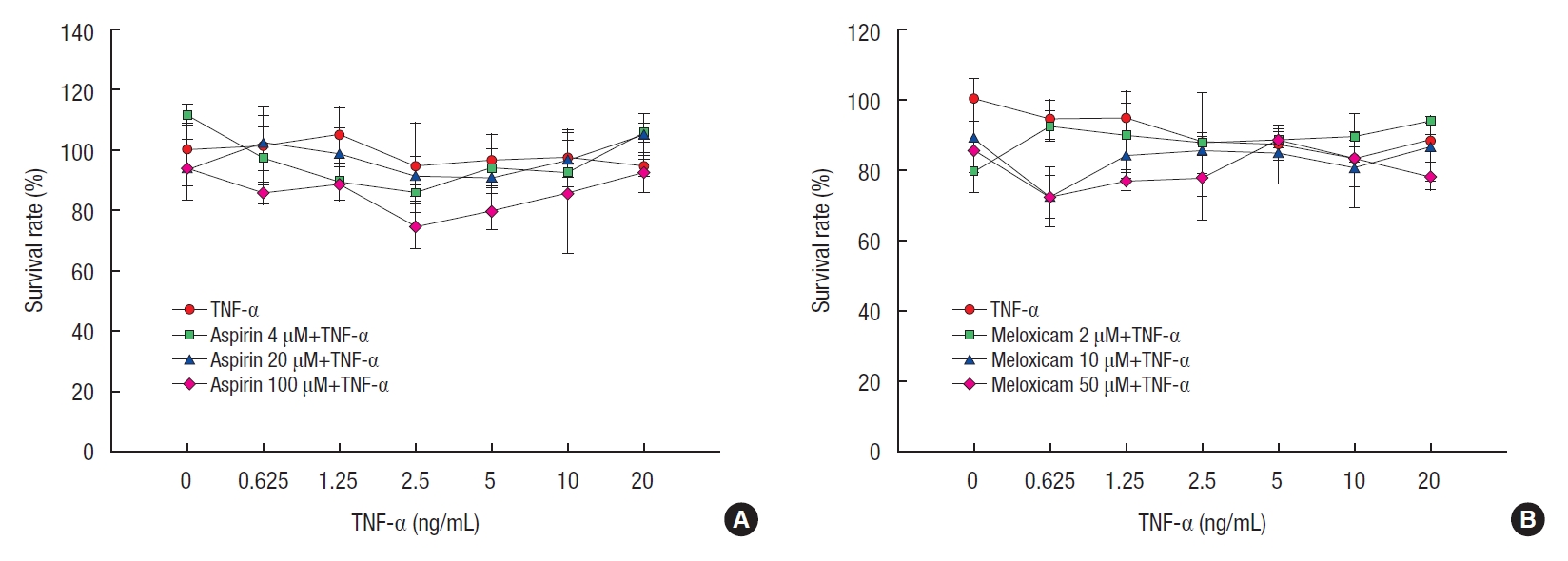
Cytotoxic effect of aspirin and meloxicam combined with TNF-╬▒ treatment in FET cells
The cytotoxic effects of aspirin, a COX inhibitor, and meloxicam, a selective COX-2 inhibitor, on cancer cells were evaluated using the MTT assay after activating FET cells upon administration of TNF-╬▒ (Fig. 5). Increases in the concentration of both aspirin and meloxicam had no impact on cell survival. These results suggest that the concentration of PGE2 does not induce cytotoxic effects.
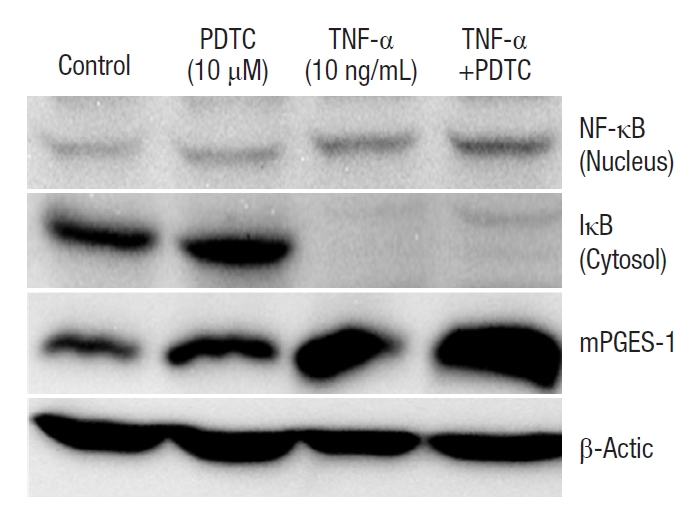
Expression of NF-╬║B and I╬║B by TNF-╬▒ and PDTC in FET cells
No change in the expression of NF-╬║B was observed even after treatment of TNF-╬▒ or PDTC, known as a selective inhibitor of NF-╬║B. However, the expression of I╬║B showed no change in PDTC treatment but decreased in TNF-╬▒ treatment. Similarly, the expression of mPGES-1 showed no change in PDTC treatment but increased in TNF-╬▒ treatment (Fig. 6). This result suggests that upregulation of mPGES-1 by stimuli of TNF-╬▒ is closely related to the effect of I╬║B rather than that of NF-╬║B.
DISCUSSION
The current study confirmed that PGE2, which is known to be involved in the progression of colon cancer, was not highly expressed in actual colon cancer tissues compared to the adjacent normal mucosa. However, PGE2 was significantly upregulated in cancer cells by an inducer, such as TNF-╬▒, and results suggest that mPGES-1 plays a crucial role in the process.
The most significant finding of the current study was the concentration of PGE2 measured directly in colorectal cancer and adjacent normal mucosal tissues. There are few such research on colorectal cancer. Ayiomamitis et al. [10] measured the concentration of PGE2 from 49 colorectal cancer patients. PGE2 was recorded at 1,550 ┬▒ 500 pg/mL in tumor tissues, and at 2,500 ┬▒ 500 pg/mL in adjacent normal mucosal tissues, showing a statistically lower concentration of PGE2 in tumor tissues than in normal ones. Rigas et al. [11] measured the concentration of various types of PG from 21 colon cancer patients, which indicated statistically significant over-expression of PGE2 in cancer tissues when compared to adjacent normal mucosa. In the current study, however, there was no difference in the PGE2 levels of 26 colorectal cancer patients (401.23 ┬▒ 188.06 pg/mL in normal mucosa, 453.10 ┬▒ 197.31 pg/mL in tumor tissues).
PGE2 has long been considered to serve an important role in the development and progression of colon cancer. Additionally, it has been shown that inhibiting the formation of PGE2 with various COX-2 inhibitors such as aspirin, sulindac, or celecoxib is effective in treating cancer progression, but the clinical use of this treatment method is limited due to side effects. Recent studies have shown that PGE2 has a role in tumor tissues and in various types of cells adjacent to the tumors, which is recognized as the concept of the tumor microenvironment (TME). TME includes macrophages, fibroblasts, myeloid-derived suppressor cells, neutrophils, lymphocytes, endothelial cells, natural killer cells, and dendritic cells, located either in or adjacent to tumor tissues. In these cells, there are multiple PGE2 receptors (EP receptor; EP1, EP2, EP3, and EP4) which are combined with PGE2 to contribute to the development and growth of tumors, serving various roles in migration, angiogenesis, and immunosuppression [12].
COX-2 is a well-known enzyme that plays a crucial role in PGE2 expression in cancers, including colon cancer. COX-2 is reported to serve as an inducible enzyme and is constitutively expressed in colon cancer and other cancers [3, 13]. A previous study carried out by the author similarly reported over-expression of COX-2 in colon cancer [6, 8]. Elevated expression of mPGES-1, an enzyme involved in the final stage of PGE2 synthesis mediated by COX-2 was reported in a previous study by the author and in those by other researchers [6, 14]. Expression of mPGES-1 is reported to be involved in the formation of polyps which appear in the earliest stage of colon cancer [7], and to have to do with a poor prognosis of the cancer [15]. As elaborated above, mPGES-1 seems to be closely related to colon cancer progression along with COX-2.
Generally, it is found that both COX-2 and mPGES-1 are upregulated upon stimulation of lipopolysaccharide or TNF-╬▒ in macrophages or fibroblasts which contributes to PGE2 expression [16, 17]. However, they seem to manifest in slightly different ways in cancer. Yoshimatsu et al. [14] reported that COX-2 and mPGES were overexpressed in colon cancer tissues, though levels of expression varied. Upon administration of chenodeoxycholate to the HCA7 colon cancer cell line, COX-2 was induced, while no effect was observed in mPGES; however, when treated with TNF-╬▒, both COX-2 and mPGES were induced, suggesting that their expression was triggered by different contributing factors [14]. Prior reports found that COX-2 and mPGES-1 were combined in colon cancer cells involved in tumorigenesis [18], but that mPGES-1was expressed coupled with COX-1in cervical cancer [19]. In the current study, COX-2 and mPGES-1 were expressed differently in colorectal cancer tissues, and mPGES-1 upregulation induced by stimulation by TNF-╬▒, a well-known phenomenon, was observed in FET colon cancer cells. Additionally, there was no change in COX-2 expression and COX-1 was slightly elevated, confirming different patterns of manifestation of COX-2 and mPGES-1.
Among a variety of enzymes related to PGE2 metabolism, the only one that showed constant expression in the current study was 15-PGDH. It was strongly expressed in adjacent normal mucosal tissues, whereas it was weakly manifested in cancer tissues. This implies that 15-PGDH functions as a tumor suppressor and has attractive potentials as a tumor suppressor gene. A study carried out by Myung et al. [8] reported that 15-PGDH acted as an in vivo tumor suppressor in the early stages of colon tumorigenesis. A previous study conducted by the author similarly reported correlations between attenuated expression of 15-PGDH and over-expression of vascular endothelial growth factor among colon cancer patients [6], which suggests 15-PGDH has a role in inhibiting cancer progression.
TNF-╬▒ is known to have tumor-promoting effects by activating various signal transduction pathways and promoting cancer growth, invasion, and metastasis [20]. NF-╬║B is reported to be involved in PGE2 upregulation triggered by TNF-╬▒. It was reported that TNF-╬▒-induced PGE2 release in human gingival fibroblast was mediated by activation of COX-2 prompted by NF-╬║B [21]. However, the current study observed that I╬║B was associated with TNF-╬▒-induced PGE2 upregulation in colon cancer cells more so than NF-╬║B. Therefore, additional studies are needed to investigate signal transduction pathways that trigger PGE2 upregulation.
The current study has limitations due to a small number of samples used to measure the PGE2 concentration which amounted only to 26 colorectal cancer tissues. Additionally, the study was limited as it examined only mPGES-1 out of the various PG synthases involved in the production of PGE2 and examined only FET cells among several types of colon cancer cells.
Considering many studies that have shown that PGE2 plays many roles in cancer progression, it is expected that the expression of PGE2 in colon cancer will be constitutively high, but only a few studies, including the authorsŌĆÖ study, show inconsistent findings. Therefore, more comprehensive studies are needed to establish the expression pattern of PGE2 in actual colon cancer tissues. Although the authorsŌĆÖ research is limited, when estimating the expression pattern of PGE2 in colon cancer based on studies using cancer cell line and actual cancer tissues and normal mucosa tissues, the expression of PGE2 is maintained at a low level in the basal state. It is presumed that PGE2 is induced and overexpressed in response to some stimuli such as cytokine, hypoxia, and chemotherapeutic agents. In addition, at this time, it is thought that mPGES-1 plays an important role as an inducible enzyme and COX-2 plays as a constitutive enzyme. However, more detailed basic research is needed to elucidate the role of these enzymes on PGE2 synthesis in colon cancer.
These results demonstrated that PGE2 can be induced by stimuli such as TNF-╬▒ and suggest that activation of mPGES-1 is more closely related than that of COX-2 in the induction of PGE2 on colon cancer.